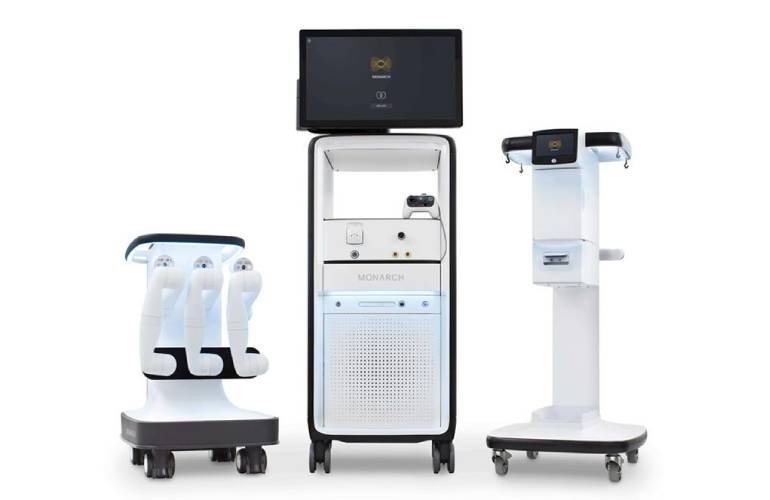
Home » Johnson & Johnson MedTech advances surgical robotics with Nvidia AI, plans 2026 launch The Monarch platform for urology leveraging AI-driven simulation with Nvidia accelerated compute. [Image courtesy of Johnson & Johnson MedTech] Johnson & Johnson MedTech (NYSE: JNJ) today announced new advances in surgical robotic technology with physical AI capabilities. The medtech giant

DeepSomatic AI: 98% accuracy in cancer mutation detection – R&D World
At its “Research to Reality” media event on Thursday, Google showcased DeepSomatic, an AI tool that accurately identifies cancer-causing genetic mutations, outperforming current methods according to a study published in Nature Biotechnology on October 16.”DeepSomatic is an AI tool that helps scientists find genetic variants in cancer cells that could enable personalized treatment,” said Yossi Matias, VP and head of Google Research, in a keynote. “It’s part of our quest to better understand the genome and to provide tools to scientists: an open-source platform that scientists can build on.””Over this decade, we’ve had DeepVariant, DeepConsensus, DeepCulture, and now DeepSomatic—all steps toward tackling cancer and other diseases,” Matias said.
Google’s Genomics Research Timeline
DeepSomatic marks the continuation of more than a decade of genomic research at Google and Alphabet. The company’s AI-driven genomics tools have contributed to landmark achievements including the first complete human genome sequence and conservation efforts for endangered species.
- 2015: Google applies deep learning to genomics, winning the 2016 PrecisionFDA Truth Challenge
- 2018: DeepVariant released—a deep learning variant caller that becomes widely adopted in genomics research
- 2022: DeepConsensus improves long-read sequencing accuracy; NIH T2T consortium completes the first truly complete human reference genome using DeepVariant
- 2023: DeepVariant and DeepConsensus contribute to the first human pangenome reference; AlphaMissense predicts disease-causing genetic variants
- 2025: DeepPolisher enhances genome assembly accuracy; AlphaGenome predicts non-coding variant effects; DeepSomatic identifies cancer mutations
Source: 10 years of genomics research at Google
In peer-reviewed testing published October 16, DeepSomatic evaluated six matched tumor-normal cell-line pairs sequenced on Illumina, PacBio HiFi and Oxford Nanopore platforms using the open CASTLE dataset (SRA BioProject PRJNA1086849). The tool consistently outperformed widely used callers against MuTect2, Strelka2, and SomaticSniper on short reads, and against ClairS on long reads.
The most significant improvements appeared in insertion-deletion (indel) detection: a challenging variant class that often causes cancer through frameshift mutations. On Illumina data, DeepSomatic achieved approximately 90% F1 score compared to 80% for the next-best tool; on PacBio HiFi sequencing, it exceeded 80% F1 versus below 50% for the comparator, a gain of 30 percentage points or more. For single-nucleotide polymorphisms on Illumina, the tool reached an F1 of 0.983 on a held-out chromosome 1 benchmark in conference results.
The tool works across three major DNA sequencing platforms: Illumina, PacBio and Oxford Nanopore. It can analyze damaged tissue samples commonly used in clinical settings, addressing a key bottleneck in precision oncology workflows.
At the research event in Mountain View, researchers explained the tool using a puzzle analogy: identifying cancer mutations is like having 1,000 sets of puzzle pieces and searching for the ones that don’t quite match the picture on the box, which represents the reference human genome. DeepSomatic distinguishes somatic mutations (genetic changes that drive cancer) from inherited variants, a critical distinction for precision oncology treatment decisions.
Technical implementation and clinical feasibility
Unlike traditional variant callers that rely primarily on statistical models, DeepSomatic extends the DeepVariant pipeline by analyzing aligned sequencing reads through a convolutional neural network. The tool is trained on tumor-normal paired samples and generates “pileup image tensors” from both tumor and healthy tissue: visual representations encoding base calls, quality scores, and alignment context—then classifies variants into categories including somatic mutations, inherited variants (germline) and low-quality calls.
According to documentation in the open-source repository, the system requires standard bioinformatics inputs: aligned BAM or CRAM files with index files and a reference genome in FASTA format, and outputs standard VCF files with custom FILTER tags distinguishing somatic variants (PASS) from germline calls. This allows integration into existing clinical workflows without specialized preprocessing. The tool accepts outputs from standard aligners such as BWA and offers platform-specific models for whole-genome sequencing (WGS), whole-exome sequencing (WES), and formalin-fixed paraffin-embedded (FFPE) tissue samples across all three major sequencing platforms.
Runtime performance varies by sequencing depth and platform. According to repository metrics documented on n2-standard-96 instances (96-core, 384GB RAM), whole-exome analyses completed in approximately 15-30 minutes, while whole-genome sequencing required 3-6 hours. The tool runs on CPU-only systems without requiring GPU acceleration, though GPUs can speed up variant calling. The parallelizable architecture scales to available computing resources, making the tool practical for hospital informatics environments through batch overnight processing or cloud computing options.
Clinical validation and reproducibility
Beyond benchmark scores, the team tested DeepSomatic’s generalization to other cancer types. According to Google Research, the tool successfully identified variants in a glioblastoma sample and, working with partners at Children’s Mercy in Kansas City, analyzed eight previously sequenced pediatric leukemia samples. In the tumor-only leukemia cases, DeepSomatic identified all previously known variants plus 10 new ones, demonstrating its ability to work with tumor-only samples where matched normal tissue isn’t available.
The tool also performed well on formalin-fixed paraffin-embedded (FFPE) tissue and whole-exome sequencing data—both common in clinical archives. Performance on FFPE tissue, which comprises the majority of archived cancer pathology samples, shows reduced recall (approximately 82% versus 95% on fresh-frozen samples, according to repository benchmark metrics in metrics.md) but maintains clinical utility. The tumor-only mode cross-references population databases to filter germline variants, maintaining approximately 90% sensitivity but with reduced precision (around 77% compared to 98% in tumor-normal mode, per repository benchmarks).
The authors report F1-scores (balancing precision and recall) rather than generic accuracy metrics, and note that prospective clinical validation will still be needed before routine clinical deployment.
Google and UC Santa Cruz have released DeepSomatic’s code on GitHub under an open-source license, alongside the CASTLE benchmark dataset deposited in the NCBI Sequence Read Archive (BioProject PRJNA1086849). The availability of training code, model weights and standardized test data enables independent validation.
Related Posts